Finding Ligands
The selection of a ligand for a PROTAC molecule is not necessarily a challenging task, but can be quite detailed. The first stage in this development would be to decide between the use of a peptide-based or a small molecule-based PROTAC ligand, which both have their advantages. Peptide-based designs are those that imitate a natural protein-protein interaction with the protein of interest (POI) via a peptide. This improves a PROTACs in-vitro characteristics, including selectivity and affinity. However, it significantly hinders the functional use of the molecule in vivo. This is because these molecules are significantly too large and hydrophilic for cell permeability in oral absorption. This makes them particularly suited to IV administration, and as such are useful molecules in targeting blood disorders (1). This is why small molecule PROTACs are chosen for disorders in tissue, like breast cancer. Although these molecules are much harder to design with the same level of selectivity or affinity, they are much more useful due to their better pharmacological properties (2). As such, we must design a small molecule PROTAC ligand in this project.
In most cases, the protein that is being targeted would have some available literature on already existing drugs which can then be modified or improved, and then used as the ligand (3). This process works as there are various chemical properties that are required by a PROTAC ligand outside of binding affinity. In fact, binding affinity is not a major concern for a ligand design, as the ligand will only need to transiently bind to the protein of interest (POI) in order to activate the E3 ligase molecule. Other properties that are more important include the pharmacokinetics and pharmacodynamics of the molecule, to ensure the PROTACs will reach the targeted cells and the targeted cell compartment (4). As such, previously researched drugs are favourable ligands to design PROTACs from. Unfortunately, both our proteins of interest, ATM and POP1 have no approved drugs as of yet, and as such there are many steps required in the development of PROTAC ligands for these proteins. However, due to the state of research on the two proteins being at two very different stages, we are proposing two different methods to develop these ligands. ATM is a protein that has an established role in lots of diseases, including various types of cancer, ataxia telangiectasia and other neurodegenerative conditions (5). Due to this, there is already a list of active ligands that could be modified, up to 159 according to PHAROS, a comprehensive database that collates information about drugs and their protein targets (PHAROS).
PHAROS Results
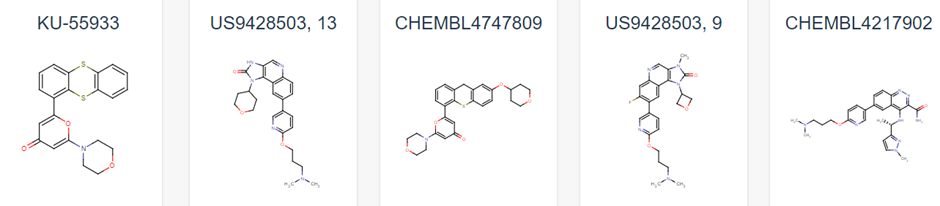
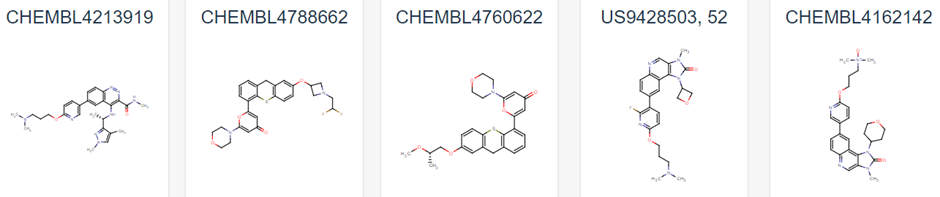
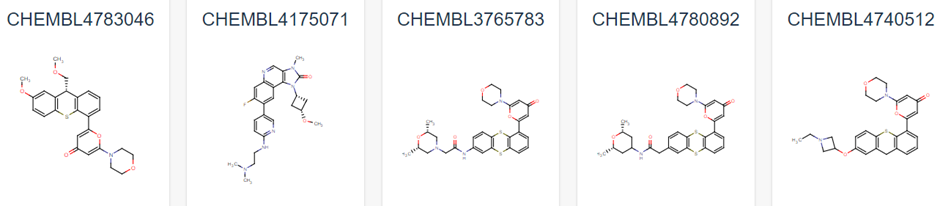
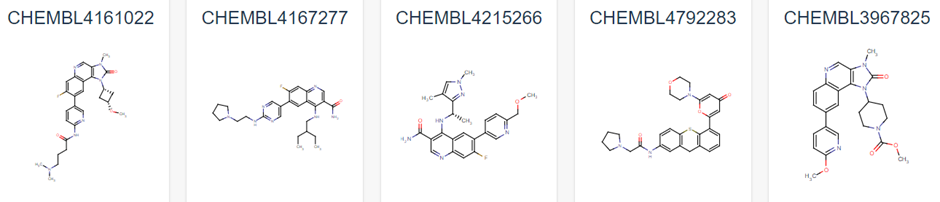
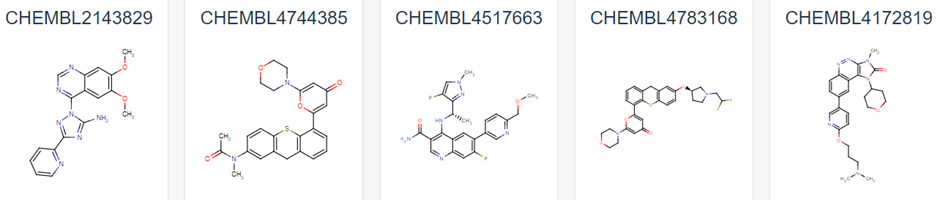
Figure 1 demonstrates some of these established chemical entities, focussing on those that are relatively selective to ATM and have favourable in vivo potency scores. Potentially, all of these could be used as ligands for the potential PROTAC molecule, but this may lead to intellectual property concerns. As can be observed in Figure 1, all have a backbone structure with various sets of aromatic rings, often with three fused together and 1 or 2 rings attached via a methyl group. This observation, as well as the whole 159 ligand list within the database opens up opportunity into rational design for a novel ligand. We could use a variety of open-source Quantitative Structure Activity Relationship (QSAR) algorithms in order to adapt these drugs. These molecules will have been optimised for their affinity to ATM in their in vivo studies, and as such we can adjust them to have better pharmacokinetic properties as affinity won’t matter as much in a PROTAC design (4). We would be focussing on aqueous solubility, cell permeability and obtaining low blood-brain-barrier permeability. As such, we suggest that the BioPPSy platform should be used in this study in order to optimise the PROTAC ligand identified, as this software focusses on these pharmacokinetic properties (6). Through this software, we propose designing a list of PROTAC ligand molecules for ATM, which will then be later validated experimentally in Aim 2.
Targeting POP1 with a PROTAC molecule is quite a different process. Due to its only recently discovered role in cancer and relatively low relevance in other diseases, POP1 still remains relatively unstudied. There are no available inhibitor structures in public databases, which makes the ligand design process more complicated. Usually, when there are no available inhibitors, the structure of the protein’s substrate is usually adapted for PROTAC design. However, POP1 does not have a small chemical substrate that can be used for this purpose, as it is an RNA binding protein (RBP), and RNA is too easily degraded within cells. However, there is a new protocol for RNA-PROTACs that are specifically designed to degrade RBPs. It involves taking the RNA sequence that the POI binds to, and replacing the phosphodiester backbone with phosphothiate linkages. Additionally, the 2’ hydroxyl group of the ribose is alkylated for better bioavailability and stability (7). As such, we must choose a sequence for the RNA molecule. It is unclear as to what part of the TERC sequence that POP1 binds to due to its only recent research, and so we propose to use a different base sequence. The sequences that we propose to validate in Aim 2 are from the P7 stem of H1 RNA. This is because the POP1 n-terminal domain has been shown to have a strong interaction with this RNA through Cryo-EM structures of Ribonuclease P (Wu et al, 2018). The sequence is as follows: 5’-GGCCAGCGAAGUG-3’. It is suggested in the RNA-PROTAC protocol that a 7 nucleotide sequence should be used, so there are multiple sequences to validate in the P7 sequence. If these do not work, sequences from P4 or CR-IV may work, but these were not chosen as P4 seems to be less available in the Ribonuclease P structure to POP1, and the concern of off-target effects if we use a highly conserved region of the RNA like CR-IV (8), but they may prove useful if studies with the P7 stem sequence do not work.
Ultimately, the design of the two PROTAC molecules for ATM and POP1 are significantly different, with one using established inhibitory ligands and a QSAR approach, and the other using a new RNA-PROTAC protocol. These approaches will provide us plenty of options to test in Aim 2, and also the opportunity to revisit these steps if the experimental methods do not yield any results. Additionally, these two methods are broad enough for us to do similar research into other proteins of interest down the line, especially as proteins that are related to the act of telomerase in cells may already have existing ligands, or may be RBPs. This protocol gives us freedom to approach both.