We first use Freeman’s betweenness centrality to naively determine protein targets (1) and visualise the local topology of the network around telomerase. These targets should be most influential in the telomerase complex’s interaction with the rest of the cell. Mainly, as a group we liked ‘betweenness’ because it allowed us to visualise the network. We hoped it would give us an idea of how close target proteins are to telomerase in what is an otherwise extremely complicated network. It however, also gives a baseline set of protein targets which we can use later as guinea pigs for our mathematical validation.
The following section details the code used to find the highest betweenness proteins (with respect to paths leading to telomerase). There is no emphasis on computational efficiency, and much of this code has been written from scratch using basic networkx commands.
A Visualisation Based on Shortest Paths
The network is read in as usual:
Setup
#~~~~~~~~~~~~~~~~~~~~~~~~~~~## PACKAGES & MODULES#~~~~~~~~~~~~~~~~~~~~~~~~~~~import networkx as nximport numpy as npimport scipy as spimport igraph as igimport matplotlib.pyplot as pltimport math import pandas as pdimport seaborn as snsfrom networkx.drawing.nx_agraph import graphviz_layoutimport pygraphviz#~~~~~~~~~~~~~~~~~~~~~~~~~~~# Beginning set-up#~~~~~~~~~~~~~~~~~~~~~~~~~~~G0 = nx.read_weighted_edgelist('../Yeast Data/4932.protein.links.v12.0.txt',comments="#",nodetype=str)threshold_score =700for edge in G0.edges: weight =list(G0.get_edge_data(edge[0],edge[1]).values())if(weight[0] <= threshold_score): G0.remove_edge(edge[0],edge[1])largest_cc =max(nx.connected_components(G0),key=len)G = G0.subgraph(largest_cc)# convert to igraph g = ig.Graph.from_networkx(G)#~~~~~~~~~~~~~~~~~~~~~~~~~~# Protein Information#~~~~~~~~~~~~~~~~~~~~~~~~~~class ProteinInfo: p_id ="ID EMPTY" p_name ="NAME EMPTY" p_size ="SIZE EMPTY" annot ="ANNOTATION EMPTY"def__init__(self, id, name, size, annot):self.p_id =idself.p_name = nameself.p_size = sizeself.annot = annotdef __print__(self):return p_name#~~~~~~~~~~~~~~~~~~~~~~~~~~~# reading in protein annotations & parse into list P of ProteinInfo objects#~~~~~~~~~~~~~~~~~~~~~~~~~~~withopen('Yeast Data/4932.protein.info.v12.0.txt', 'r') as info: lines = info.readlines()P = []i =1while i <len(lines): line = lines[i].split('\t') P.append(ProteinInfo(line[0].strip(), line[1].strip(), int(line[2].strip()), line[3].strip())) i +=1#~~~~~~~~~~~~~~~~~~~~~~~~~~~# ProteinInfo class related functions#~~~~~~~~~~~~~~~~~~~~~~~~~~~# returns ProteinInfo object from protein iddef pro_info(list, protein):for p inlist:if protein == p.p_id:return preturn"ERROR: protein not found"# returns ProteinInfo object from protein namedef pro_info_from_name(list, protein):for p inlist:if protein == p.p_name:return preturn"ERROR: protein not found"# returns list of proteins that contain the given string in their annotationdef find_p(list, string): find_poi = []for p inlist:if p.annot.find(string) !=-1: find_poi.append(p.p_id)return find_poi#~~~~~~~~~~~~~~~~~~~~~~~~~~~## Telomerase Reference Material:#~~~~~~~~~~~~~~~~~~~~~~~~~~~G # NetworkX networkg # igraph networkP # protein annotations# Target Proteinspop1 = pro_info(P, '4932.YNL221C')tel1 = pro_info(P, '4932.YBL088C')# Telomerase Proteinsest1 = pro_info(P, '4932.YLR233C')est2 = pro_info(P, '4932.YLR318W')est3 = pro_info(P, '4932.YIL009C-A')# lists for accessing poi = [pop1, tel1, est1, est2, est3]poi_id = ['4932.YNL221C', '4932.YBL088C', '4932.YLR233C', '4932.YLR318W', '4932.YIL009C-A']tel_id = ['4932.YLR233C', '4932.YLR318W', '4932.YIL009C-A'];target_id = ['4932.YNL221C', '4932.YBL088C']T ='telomerase'
The key point is that we collapse the 3 telomerase proteins into a single protein. This allows us to find the shortest paths to the telomerase complex without having to take the minimum of all paths to the individual telomerase proteins, which improves computational efficiency 3-fold. We achieve this through the networkx commands contracted_nodes and relabel_nodes, and relabel the conjoin telomerase complex ‘telomerase’.
#~~~~~~~~~~~~~~~~~~~~~~~~~~~# Conjoin telomerase into single entity:#~~~~~~~~~~~~~~~~~~~~~~~~~~~G = nx.contracted_nodes(G,tel_id[1], tel_id[0], self_loops=True)G = nx.contracted_nodes(G,tel_id[1], tel_id[2], self_loops=True)# relabel tel_id[1] node as `telomerase' for ease of referencemapping = {'4932.YLR318W':T}G = nx.relabel_nodes(G, mapping)
First, we generate a color-map for the final network:
#~~~~~~~~~~~~~~~~~~~~~~~~~~~def get_tel_color_map(G, target_id):# Generate colour map to visualise the telomerase and key proteins for a given graph. color_map = []for node in G.nodes():if node == T: color_map.append('red')elif node in target_id: color_map.append('orange')else: color_map.append('black')return color_map
We then find the length of the shortest paths to telomerase:
2.1 Get Shortest Path Lengths
###### 2.1: Get the "shortest-path-lengths" (SPL) to a telomerase protein (and keep the shortest one of the three).tel_SPL = {};for node in G.nodes(): L = nx.shortest_path_length(G, T, node); tel_SPL[node] = L
This allows us to obtain a subgraph of all nodes with shortest path length k away from telomerase:
2.2 Get subgraph of the k-closest nodes.
###### 2.2: Get the graph of the closest (k distance) proteins based on SPL.def k_closest_to_tel(G, tel_SPL, k): k_tel = nx.Graph(G);# make a copy to unfreeze graph.for node in G.nodes():if tel_SPL[node] > k: k_tel.remove_node(node)print("Number of nodes: ", k_tel.number_of_nodes()) # for interest.return k_tel
This subgraph is then fed into the following function to remove all paths other than those involved in a shortest path to telomerase.
2.3 Remove all non-shortest path edges
###### 2.3: Remove edges in the graph that are not part of shortest paths to telomerase.def keep_shortest_path_edges(k_tel, tel_id, tel_com_SPL): k_tel_SPs = nx.Graph(k_tel) # create copy to remove edges from.### a) iterate through nodes (and tel proteins), and generate list of edges that appear in shortest paths SP_edges =set({})for node in k_tel.nodes(): SP_nodes = nx.all_shortest_paths(k_tel, source=T, target=node, weight=None, method='dijkstra')for path in SP_nodes:iflen(path) == tel_SPL[node] +1: # so get shortest# only count the shortest paths to telomerase proteins (no unneccessary p-p paths)for i inrange(0, len(path) -1): start = path[i]; stop = path[i+1];# add both edges both because might be in different order in the graph edge list... SP_edges.add((start, stop)) SP_edges.add((stop, start))### b) all edges in the graph at the moment: all_edges =set(nx.edges(k_tel));### c) take a set-wise difference of the two: remove_edges = all_edges.difference(SP_edges)### d) remove unwanted edges from the graph:for u, v in remove_edges: k_tel_SPs.remove_edge(u, v)return k_tel_SPs
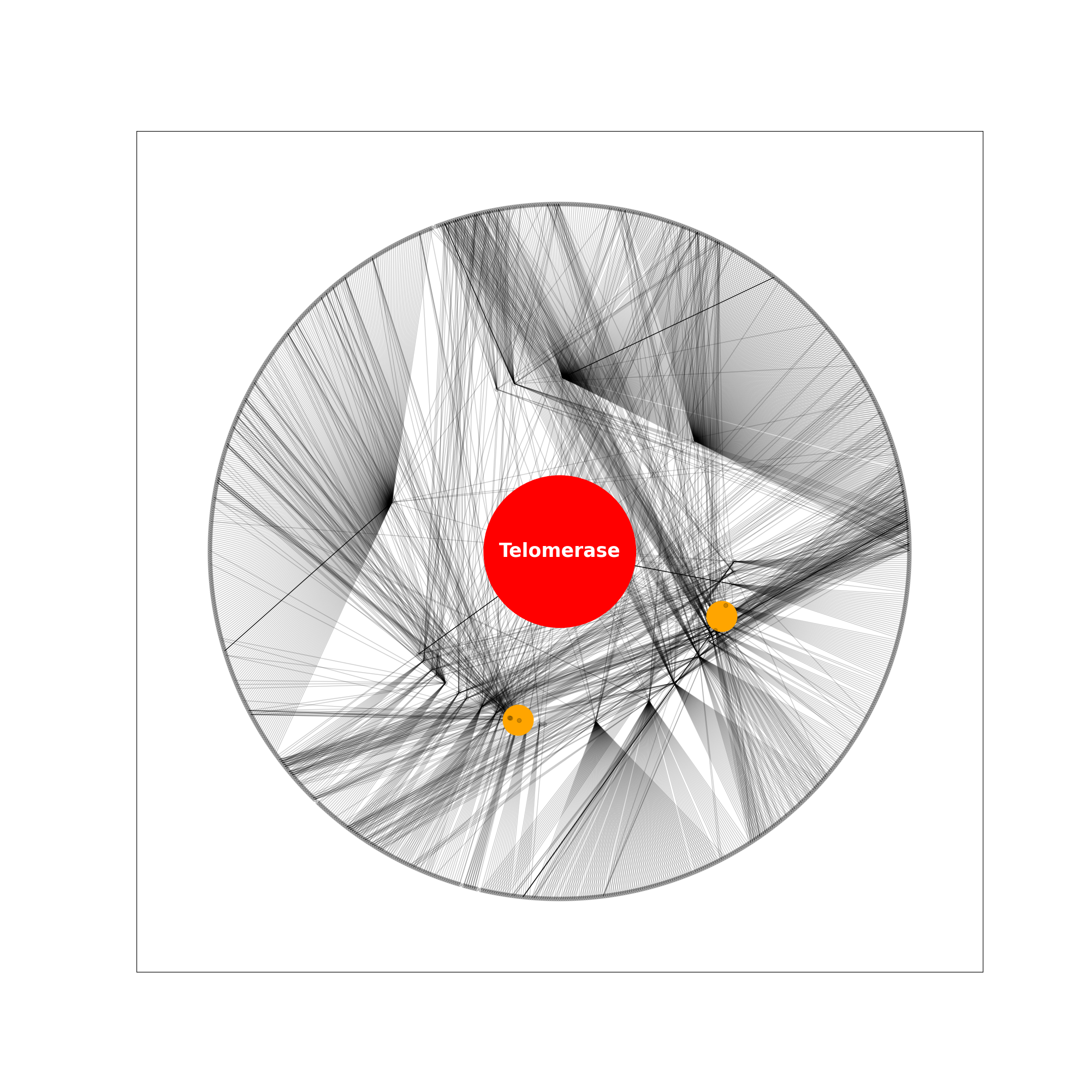
For example, the following figure is obtained showing all shortest paths nodes that are a shortest-path-distance 2 away from telomerase, with telomerase in the centre and nodes POP1 and ATM labelled in orange. The network is visualised using the graphviz_layout layout ‘twopi’ with telomerase at the centre. Nodes that are distance 1 away from telomerase are in the central ring, and nodes distance 2 away are in the outer ring.
An example for k = 2.
# Again, remove all paths that are not part of the shortest paths.fig = plt.figure(figsize=(50,50))k =2k_tel_G = k_closest_to_tel(G, tel_SPL, k)k_tel_G_SPs = keep_shortest_path_edges(k_tel_G, tel_id, tel_SPL);# set-up:fig = plt.figure(figsize=(20,20))color_map = get_tel_color_map(k_tel_G_SPs, tel_id, target_id)# position:pos = graphviz_layout(k_tel_G_SPs, 'twopi', root=T) # <------ this is by far the nicest to visualise!node_sizes = []alphas = []for node in k_tel_G_SPs.nodes():if node == T: node_sizes.append(40000) alphas.append(1)elif node in target_id: node_sizes.append(1600) alphas.append(1)else: node_sizes.append(30) alphas.append(0.2)nx.draw_networkx(k_tel_G_SPs, pos=pos, node_size=node_sizes, node_color = color_map, with_labels=False, alpha=alphas)nx.draw_networkx(k_tel_G_SPs, pos=pos, node_size=40000, edgelist=[], edge_color='k', nodelist=[T], labels = {T:'Telomerase'}, node_color='red', font_color='white', font_size=25, font_weight='bold')
Visualisation of proteins distance 2 away from telomerase.
Calculating Betweenness
Here, we calculate a betweenness-like measure for each protein. It counts the number of shortest paths directed towards the telomerase protein, travelling through a given protein. We call this the telomerase betweenness. Again, much of the computation is brute-force. First, we obtain sub-dictionaries of proteins which are distance k away from telomerase, and greater or equal to distance k away from telomerase:
Sub-dictionaries for proteins
def k_dictionary(tel_SPL, k):# all proteins with shortest path length k distance awayreturndict(filter(lambda path_lenth: path_lenth[1] > k-1and path_lenth[1] < k+1, tel_SPL.items()))def greater_than_k_dictionary(tel_SPL, k):# all proteins with shortest path length k or greater distance awayreturndict(filter(lambda path_lenth: path_lenth[1] > k-1, tel_SPL.items()))
This allows us to find the telomerase betweenness of all proteins that are k shortest distance away from telomerase (we refer to the set of these proteins as a k shell). We do so by iterating over all shortest paths directed towards telomerase that start at proteins which are further than k distance away from telomerase, and count the number of times proteins in the k shell appear in these paths:
Finding the telomerase betweenness of a k-shell
def k_shell_betweenness_to_telomerase(tel_SPL, k):# find the betweenness of all proteins k shortest path distance away from telomerase # iterate over proteins greater than k distance away and calculate tel_SPL_k = k_dictionary(tel_SPL, k) greater_than_tel_SPL_k = greater_than_k_dictionary(tel_SPL, k) shortest_paths = []for p in greater_than_tel_SPL_k.keys(): shortest_paths = shortest_paths +list(nx.all_shortest_paths(G, source=T, target=p)); betweenness = {};for interest in tel_SPL_k.keys(): btw =0;for path in shortest_paths:for i inrange(1,len(path)):if path[i] == interest: btw +=1 betweenness[interest] = btwreturn betweenness
Results
From this we obtain the following proteins that have the highest telomerase betweenness:
Freeman LC. A set of measures of centrality based on betweenness. Sociometry [Internet]. 1977 [cited 2023 Oct 24];40(1):35–41. Available from: http://www.jstor.org/stable/3033543