Evaluation of Ligands and Integration into PROTACs
With a set of ligands identified that have the potential to have excellent binding affinities for the proteins of interest, they must all be quantitatively validated and compared in order to find the best in class ligand for each of the target proteins. The best ligands can then be integrated into the proposed protein therapeutic. Hence to accomplish this, each target protein will have to be expressed and purified, and then tested against each of the potential ligands. The methods used to conduct this are reported below.
Overexpression and Purification
POP1
Human POP1 has not been successfully expressed and purified in the literature. However, the yeast homolog of human POP1 has been (1), and hence for this project, the yeast homolog will be overexpressed and purified for use in binding assays with the suggested ligands. Purification of the POP1 in isolation however has been unsuccessful, and it must be co-expressed with POP4 for solubility, and then isolated separately.
Overexpression
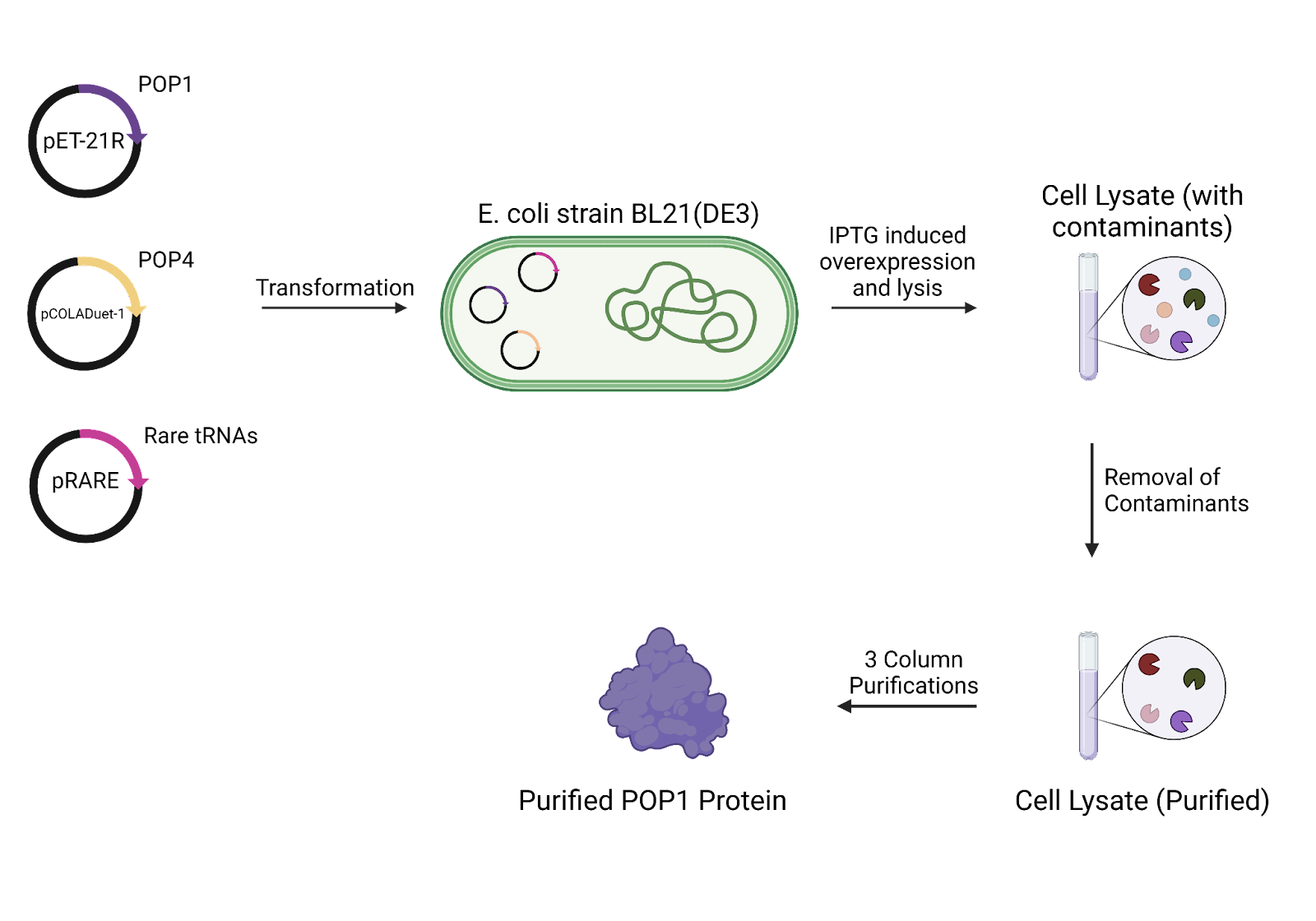
The POP1 sequence will be amplified using PCR from S. cerevisiae strain W303 before insertion into a pET-21R plasmid. An artificial POP4 will then also be cloned into a digested pCOLADuet-1 plasmid. E. coli strain BL21(DE3) will then be transformed with the pET-21R-based POP1 plasmid, the pCOLADuet-1-based POP4 plasmid and the pRARE plasmid (which contains a number of rare E. coli tRNA codons). The transformed cells will have over-expression induced using IPTG.
Purification
Cells will be pelleted using centrifugation lysed in a lysis buffer. The lysate will then be clarified using sonication and further centrifugation. Contaminants such as nucleic acids will be removed using Tween-20 to solubilise the protein and polyethylenimine to precipitate nucleic acids and other contaminants. Finally the protein of interest will be precipitated using ammonium sulfate and will be collected using centrifugation.
This pellet will be dissolved in buffer before being incubated with Ni-NTA resin. This mixture will then be purified using a Ni-NTA resin column, wherein POP1 will stick to the resin while POP4 will elute quickly. The buffer will be supplemented with Na-imidazole to elute POP1. This purified eluent will then be further purified using a second SP-Sepharose column. Finally this further purified eluent will be fractionated using a Superdex 200 gel-filtration column, yielding the purified POP1 protein.
Analysis
Protein yield will be determined using a Nanodrop spectrophotometer through measurements of absorbance at 280 nm and comparison to the extinction coefficient of Pop1 (1.11 × 105 M−1cm−1) which was determined using the ExPASy ProtParam Tool. Protein purity will be confirmed through mass spectrometry analysis.
Expected Outcome
The protocol that this proposal is based on achieved a final protein yield of 350 µg of POP1 with a high purity. The paper specifically reports that their sample was nucleic acid and RNase-free. Hence there is a strong precedent that this procedure will be able to yield large quantities of POP1 in a high enough purity to be used for further analysis.
ATM
A previously conducted paper has reported successfully expressing an amino-terminal FLAG-epitope tagged ATM protein within HEK 293T cells (2). This same paper reported successful purification using anti-FLAG affinity chromatography with a reasonably high yield and purity. Hence this project will use the same methodology.
Overexpression
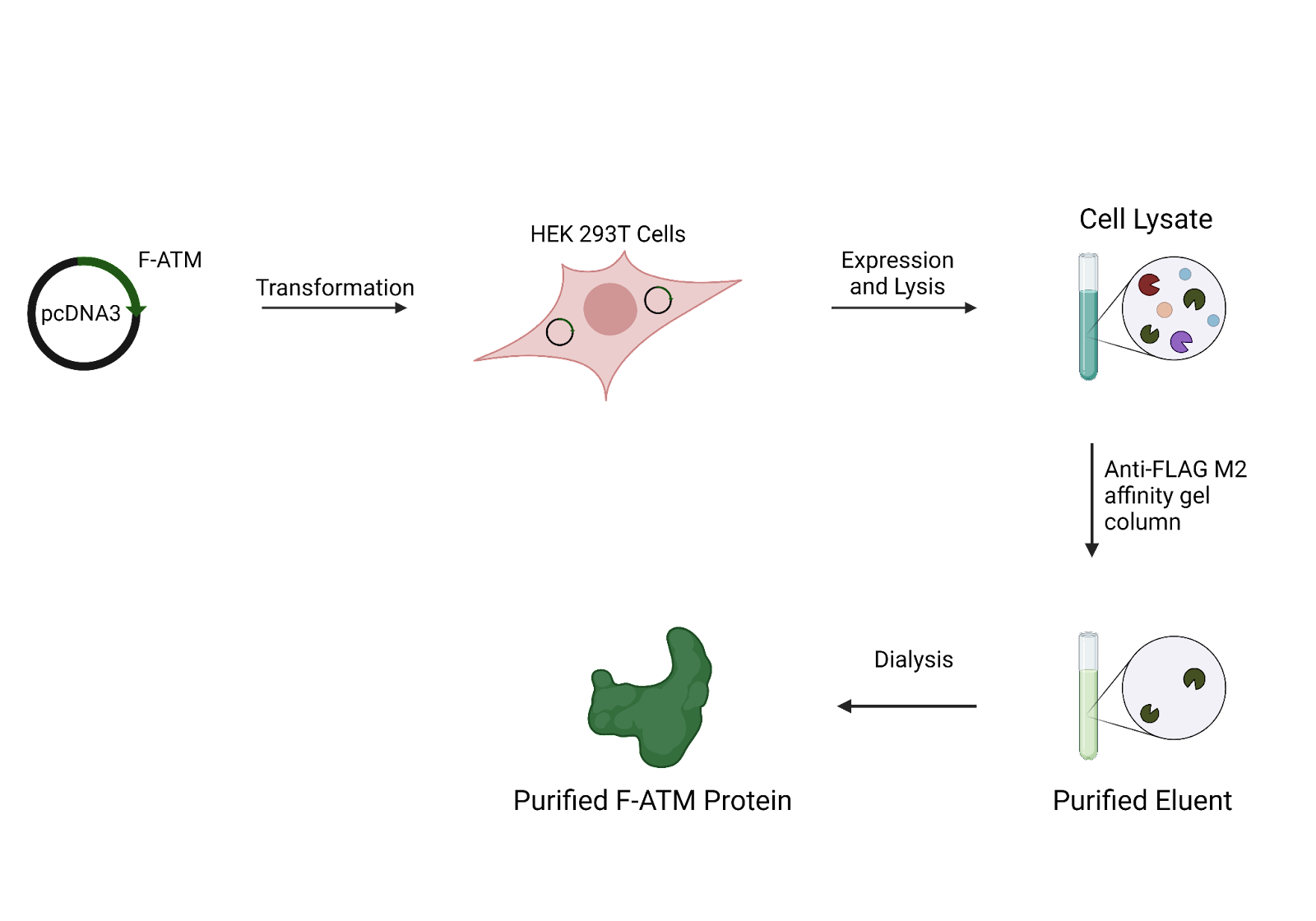
pcDNA3-F-ATM plasmids will be used for overexpression of ATM. To overcome the instability of the ATM cDNA constructs, the plasmid will be propagated within the SURE strain of E Coli. Plasmid DNA will be prepared using a Giga Kit. This plasmid DNA will be transformed into HEK 293T cells, which will be harvested using trypsin followed by centrifugation.
Purification
For purification, the cell pellets will be lysed in a lysis buffer, followed by clarification using centrifugation. This clarified lysate will then be purified using an anti-FLAG M2 affinity gel column. ATM will be eluted with FLAG peptide dissolved in buffer. The eluate will then be concentrated before dialysis which will yield the purified ATM protein.
Analysis
The successful purification of ATM can be confirmed through comparison of the purified protein to the whole cell lysate using immunoblotting. The whole cell lysate can be analysed using immunoblotting with A16.35 mAbs. The purified ATM protein can be analysed using immunoblotting with A16.35 mAbs followed by staining with Bio-Safe Coomassie.
Expected Outcome
The research paper that this methodology is based on purified 50 µg of very pure sample of F-ATM which was confirmed through immunoblotting and SDS-page analysis of the purified samples (2). Hence this methodology is expected to yield high purity ATM protein for this project.
Testing Identified Ligands
Isothermal Titration Calorimetry
These purified proteins will then be tested against the identified ligands using isothermal titration calorimetry in order to quantitatively determine their binding affinities. The methodology has been based on papers describing ITC protocols for binding assays with other nuclear proteins (4)

Method
Before implementation into a PROTAC molecule, the binding affinity of the ligands to the target proteins POP1 and ATM will be quantitatively determined using isothermal titration calorimetry (ITC). Solutions of both the proteins of interest and their small molecule ligands will be created in HEPES buffer for analysis. During the ITC assay, each small molecule ligand will be titrated against their respective protein of interest. The titration experiment will be monitored using a MicroCal iTC200 system at 37 oC.
Analysis
The resultant ITC curves are going to be analysed using a simple single-binding site model with ITC data analysis software such as origin. This will reveal the binding affinity (KA) of each of the tested ligands.
Expected Outcome
Through comparison of the binding affinities of each of the tested ligands, this will provide quantitative data on which of the identified small molecules is best able to bind to the target proteins. The best ligands can then be utilised within the proposed PROTAC therapeutic.
However there is undoubtedly a limitation in this methodology with relation to testing ligands for POP1. Due to a lack of protocols within the literature for the expression and purification of human POP1, a protocol for yeast POP1 has been proposed instead. While these proteins are homologs and hence a strong binding affinity for one could indicate a high binding affinity for the other, this is not a perfect methodology, with the potential for false positives and negatives. However, later testing of the resultant PROTACs in cell cultures will reveal if either of these are the case.
PROTAC Design
The ligands identified to have the best binding affinities for their target proteins can now be integrated into a PROTAC therapeutic.
There has been previous work done on using PROTACs to degrade the TRF1 and TRF2 proteins that form part of the shelterin complex (5). The shelterin complex has important roles in telomere protection and length regulation, and hence a PROTAC successfully able to target proteins in this complex would likely be effective at targeting ATM and POP1. Specifically, this past research has demonstrated that this PROTAC can enter and degrade proteins within the nucleus. Hence this previously reported PROTAC will serve as a framework for the PROTAC suggested in this proposal.