Biological Validation
In order to ensure that these proteins provided by the mathematical methods are genuine targets, it is important to place these proteins in their biological contexts. As seen in Table 1, the proteins with the highest mathematical scores were assessed for their subcellular location and their function. Those that were localised within the nucleus, as telomerase is, and had roles in DNA repair or the cell cycle were chosen for further study (highlighted green), as these functions establish links to telomerase. It was also essential for these proteins to have human homologs. The other proteins listed were discounted either for incongruent localization, or as they have essential cell function roles, like ribosomal subunits. These proteins are incredibly involved in a variety of cell pathways, and as such, are not promising cancer treatment targets.
Assessment of New Targets
Table 1. Initial MATH proteins proposed to the BCMB team. Assessment of the biological context of putative protein targets informed those selected for assessment in a yeast CRISPR/Cas9 knockout assay. Selected proteins are highlighted in green.
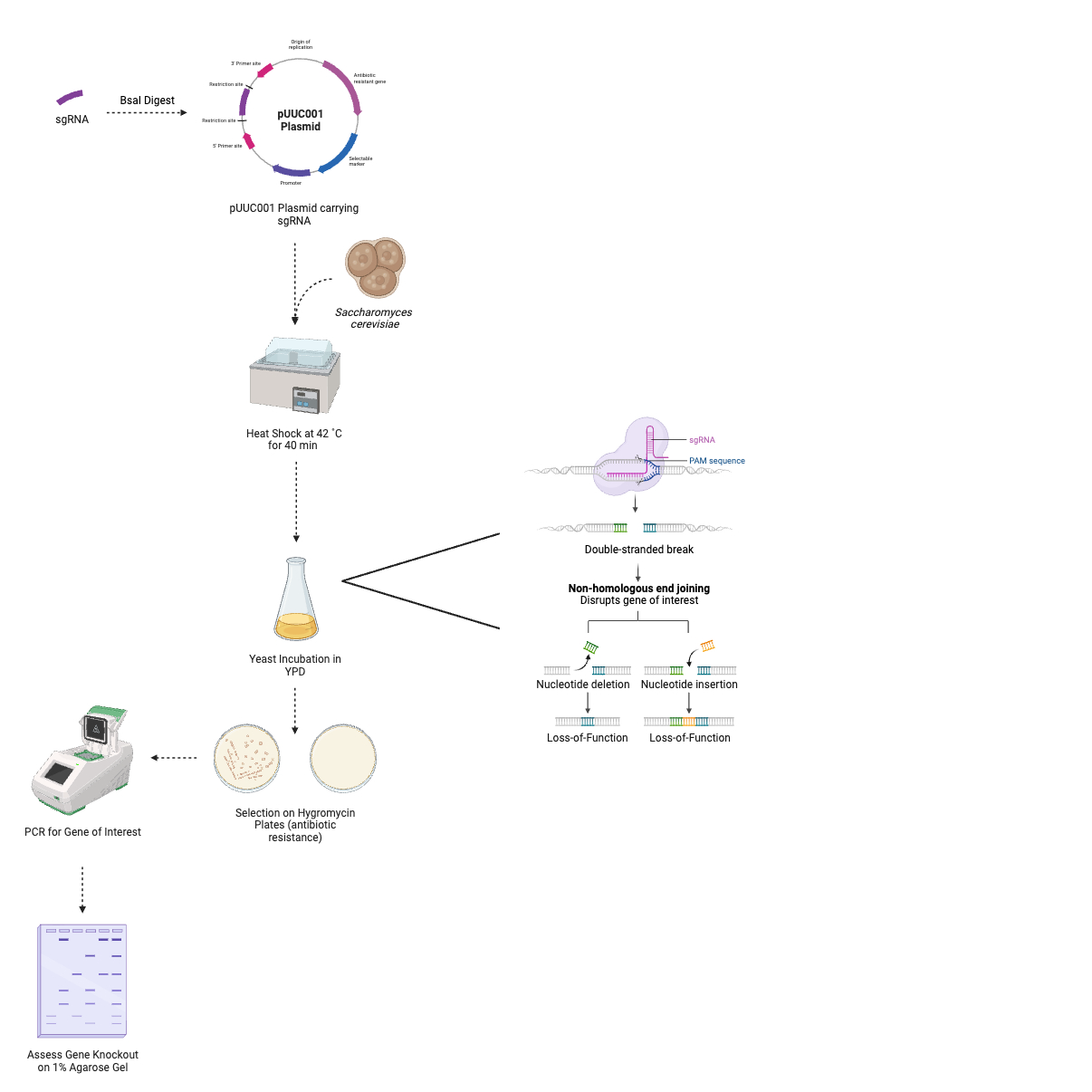
We intend to assess the pharmacological use of these proteins with a yeast viability assay of CRISPR knockouts of these proteins. Saccharomyces cerevisiae is an adaptable, simple, eukaryotic model organism to use CRISPR in (1). Our nominal Saccharomyces cerevisiae strain, BY4741, is best for use due to its 4 added CRISPR-addressable landing pads. These are places in which the genome has been modified for efficient genome manipulation (2). The CRISPR system consists of two key components: a Cas9 endonuclease and a guide RNA (gRNA). The guide RNA directs the Cas9 to the specific target sequence, which will change with each protein being knocked out. Once the Cas9 is recruited to the specific region, it will “cut” the DNA to produce a double-stranded break at a point designated by the Protospacer Adjacent Motif (PAM). This break will then be addressed by the cell’s error prone repair mechanism, non-homologous end-joining (NHEJ). As a result, in the case of a successful edit, the protein of interest’s gene will undergo loss-of-function insertions or deletions, or a premature stop codon will emerge. When this occurs, the protein becomes non-functional or cannot be expressed, thus “knocking out” the target gene (3).
By target gene knockout, we aim to find a target that will not entirely kill the yeast cells. This is to make sure that inducing degradation of these proteins in human cells will also not kill the healthy, non-cancerous cells that are exposed to our drug. The effect of knocking out these proteins on telomerase will then be assessed with a terminal restriction fragment analysis, like that described in Aim 2 – Initial Validation. Ideally, these results will demonstrate reduced telomere size. If this is occurs, these targets can then be used in the methods described in Aims 1, 2, and 3 to design and validate the respective PROTAC molecules for chemotherapeutics.
Most of the lead proteins selected from Table 1 have been chosen from the Betweenness and PCC mathematical methods as well as one obtained from non-hub proteins. If these proteins prove to be valid biological targets through our experimental validation, this will go on to testify that these mathematical methods are particularly efficient at providing these novel targets. The targets that we have chosen include MRE11, RAD51, and RAD52. All three of these are critical proteins in the double strand break repair response by homologous recombination (4). Misapplication of this pathway to telomeres has been linked to oncogenesis previously, but a CRISPR knockout experiment is needed to assess directly whether the lack of protein will cause a depletion in the telomeres. Current treatment strategies targeting these proteins are limited to small molecule inhibitors (5) and as such, developing a PROTAC for degradation of the proteins may lead to better clinical results.
RFA3 is a slightly different case. It is a subunit of the Replication Factor Protein that has major roles in cell replication, mostly in DNA recombination. Previously, a different subunit of the complex, RFA2 has been shown as having a major role in telomerase regulation, where truncation of its N-terminal region has caused severe telomere shortening (6). This suggests that RFA3 may also have a role in maintaining high telomerase activity, which should be examined in more detail. As such, a CRISPR knockout of this protein is a good place to start to investigate whether this is also a good treatment target to reduce telomerase action.
Methods
sgRNA Design
Prior to the experimental portion of CRISPR protein knockdown, initial in silico analysis and planning must occur. Because the guide RNA determines the site at which the Cas9 induces the double-stranded break (DSB), it is important that the sequence maps uniquely to the genome at the gene of interest. If the sgRNA were to guide the Cas9 to multiple locations throughout the genome, a number of different proteins could undergo loss-of-functions (7). This would certainly interfere with the validity and reliability of our experimental findings.
CRISPOR is a publicly available bioinformatic software that aids in sgRNA design (8). CRISPOR requires input of an exon sequence from the protein of interest. From this information, the algorithm designs optimal sgRNAs for use in a CRISPR/Cas9 system. Importantly, this software considers the sequence specificity to the rest of the genome, predicted editing efficiency, predicted outcome of the edit, and predicted any off-target cuts (8). See Table 2 for sgRNAs designed for knockout of the aforementioned target proteins with a Cas9 endonuclease.
Akhmetov and colleagues (2018) advise the design of multiple gRNAs for each POI knockout, as it enhances the likelihood of a successful experiment. For this reason, three putative sgRNAs will be designed then tested for each protein of interest (9). The ThermoFisher Scientific Custom DNA Oligo Synthesis Service will be used to produce gRNAs for each knockout experiment. For more details, refer to Table 2.
Plasmid Preparation
A pUUC001 plasmid (AddGene plasmid #124451) will be used to drive expression of our CRISPR system components (Figure 1). This plasmid is optimal for use of co-expression of both the Cas9 endonuclease and a guide RNA in a range of species, including Saccharomyces cerevisiae (10). This plasmid has been selected for its ease of use in terms of gRNA cloning into the plasmid in a single-tube reaction. Golden gate cloning allows for directional assembly of the plasmid with the gRNA. BsaI endonuclease cut sites flank the gRNA cloning site on the pUUC001 plasmid. The cut site is uniquely non-palindromic, meaning the same endonuclease can be used at two sites, without the risk of insertion of the gRNA into the plasmid in the incorrect orientation (11).
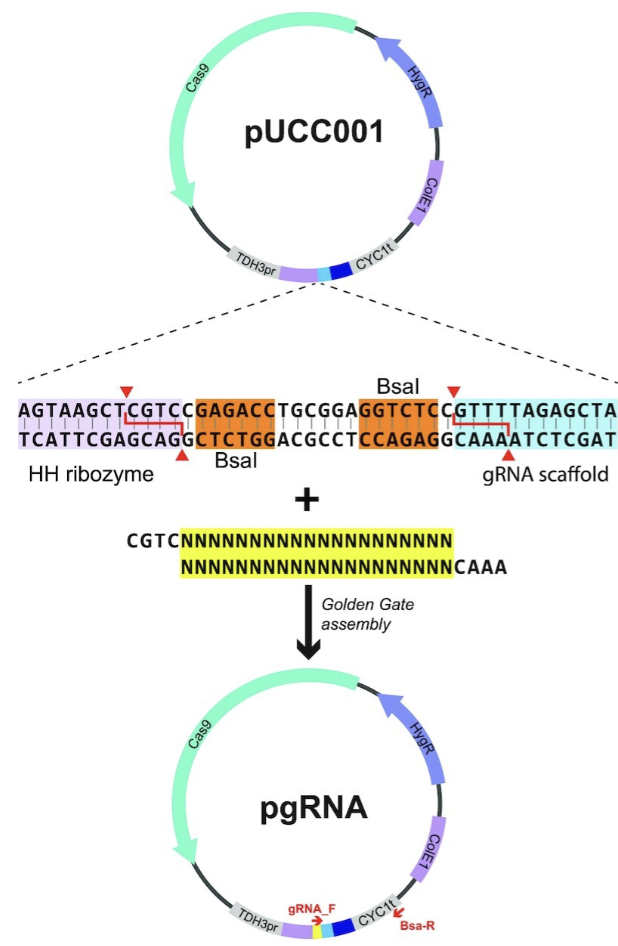
Single guide RNA constructs will be cloned into plasmids prior to yeast transformation. For this reason, the gRNAs must be ordered with the correct oligos at their 5’ ends to successfully anneal to the pUCC001 plasmid overhangs. The forward oligo has sequence 5’-CGTC-3’, and the reverse sequence is 5’-AAAC’3’.
Table 2. Target proteins from our mathematical colleagues’ investigations, and putative single guide RNA sequences designed by CRISPOR (8). Sequences also contain the forward and reverse oligonucleotide sequences to enable Golden Gate annealing into the pUCC001 plasmid.Transfection into Saccharomyces cerevisiae
pUCC001 plasmids will be purchased in E. coli culture John Morrissey Lab via AddGene. Cultures will be grown in LB and ampicillin at 37 ˚C. Plasmids will be extracted using a Gene-Jet Miniprep Kit from ThermoFisher Scientific. The complementary gRNA inserts will be added to the plasmid sample solution in sterile water. A restriction digest will be carried out with BsaI endonuclease. Equal volumes of gRNA inserts will incubated with plasmid solution in sterilised water. Complementary sequences will anneal to, thus closing the breaks and overhangs in the plasmid, and producing a pUCC001 plasmid carrying the gRNA, with expression driven by flanking promoters.
The transformation of Saccharomyces cerevisiae colonies with pUCC001 plasmids carrying gRNA will be carried out in a series of simple steps. A single colony from a plate containing growth medium will be selected, then inoculated in YPD broth overnight. The transformation mix (50% PEG 3350, lithium acetate, carrier DNA) and pUCC001 plasmid will be added to yeast colonies of uniform concentrations, quantified by optical density at 600 nm. Cells will be heat-shocked at 42 ˚C in a water bath for 40 minutes. This encourages yeast uptake of the plasmids. Following this, cells will be left for incubation at 30 ˚C. Cells will then be centrifuged, and then resuspended in YPD media. Transformed yeast cultures will be plated onto YPD + hygromycin plates. Hygromycin is an antibiotic that would usually not allow the growth of yeast on plates. However, the pUCC001 plasmid encodes a gene for resistance to hygromycin. This means that only yeast cells that have undergone successful plasmid transformation will grow. The plates will be left to dry, then incubated 30 ˚C for 3-5 days, or until colonies appear (10).
Next, colonies that have successfully undergone a gene edit will be detected. This will also allow for quantification of the efficiency of each CRISPR/Cas9 system. 10 colonies per plate will be selected at random for assessment by polymerase chain reaction (PCR). Colonies will be picked and inoculated in sterile water. A thermocycler will be used to quantify the presence of the gene of interest in the sample. A OneTaq 2X MasterMix from New England Biolabs will be used, alongside forward and reverse primers for the gene of interest. The reaction will then be run on a 1% agarose gel and stained to check for PCR products. Gene deletion will result in no amplification of the gene of interest, and therefore no band of gel at the respective sequence length (bp) (10).
Analysis
It is expected that the efficiency of the CRISPR/Cas9 system described above will be approximately 50% (10). Lethal gene knockouts cannot result in any growth of Saccharomyces cerevisiae colonies on YPD + hygromycin plates that have undergone successful loss-of-function edits (12). To draw conclusions from the experiment, it must be validated that the only colonies grown on the plate were unedited. Taking 10 samples that show unsuccessful gene knockout should give sufficient statistical power to a result that demonstrates an embryonic lethal knockout (10). The proteins that give this result will not be appropriate for use at PROTAC targets in a Homo sapiens system for chemotherapeutic use, as they will have shown to have lethal knockout.
Yeast growth of colonies that have undergone successful loss-of-function in the gene of interest will demonstrate that knockout of the protein of interest is not lethal. PCR results for colonies should show no band at the gene of interest, relative to an untreated control. This result will demonstrate that the protein of interest is not lethal when knocked out. The proteins that show this result will be appropriate for use as therapeutic PROTAC targets through our proposed approach.
Terminal Restriction Fragment Analysis
The terminal restriction fragment analysis will be performed identical to that seen within Aim 2 - Experimental Evaluation of PROTACs. The predicted results of these knockouts will be similar to those predicted for the successful PROTAC molecules, as both would theoretically shorten the telomeres. Assessment of telomere length in POI-knockout organisms will allow for assessment of whether the target proteins are essential for telomerase activity. A suitable protein target for therapeutic use will show a reduction in telomere length in response to knockout.
Future Research Applications
To summarise, the mathematically identified novel targets have been validated by searching through the available literature, and those chosen from the list will be experimentally identified by creating yeast knockouts of these genes via CRISPR/Cas9 gene editing. A yeast viability assay will be performed on the colonies grown from these cells, and those that have proven to not be lethal knockouts will be assessed for telomere length by a terminal restriction fragment analysis. From this, we hope to identify new proteins possibly implicated in telomerase activity and aim to put these proteins through the same iterative process described in Aims 1 to 3, to develop PROTACs targeting these new proteins for novel cancer treatment.